Estrés oxidativo, péptido β–amiloide y enfermedad de Alzheimer - Natalia Manzano–León y Jaime Mas–Oliva*
Estrés oxidativo, péptido β–amiloide y enfermedad de Alzheimer - Natalia Manzano–León y Jaime Mas–Oliva*
Gaceta médica de México
versión impresa ISSN 0016-3813
Gac. Méd. Méx v.142 n.3 México may./jun. 2006
Artículo de revisión
Estrés oxidativo, péptido β–amiloide y enfermedad de Alzheimer
Oxidate stress β–amiloide peptide and Alzheimer's disease
Natalia Manzano–León y Jaime Mas–Oliva*
Recibido en su versión modificada: 13 de enero de 2006
Aceptado: 27 de enero de 2006
Aceptado: 27 de enero de 2006
*Correspondencia y solicitud de sobretiros:
Jaime Mas–Oliva,
Instituto de Fisiología Celular, UNAM. Apdo. postal 70–243,
04510 México, D.F.
Tel: 5622 5584,
correo electrónico: jmas@ifc.unam.mx
Jaime Mas–Oliva,
Instituto de Fisiología Celular, UNAM. Apdo. postal 70–243,
04510 México, D.F.
Tel: 5622 5584,
correo electrónico: jmas@ifc.unam.mx
Resumen
La enfermedad de Alzheimer es la causa más común de demencia en la población de edad avanzada. Una de las características histopatológicas de esta enfermedad es la formación de placas seniles, cuyo componente proteínico es el péptido β–amiloide (Aβ) ensuforma insoluble. Este péptido se produce normalmente en forma monomérica soluble y circula en concentraciones bajas en el líquido cefalorraquídeo y sangre. En concentraciones fisiológicas actúa como factor neurotrófico y neuroprotector, sin embargo con el envejecimiento y sobre todo en la enfermedad de Alzheimer se acumula, forma fibrillas insolubles y causa neurotoxicidad.
La toxicidad del Aβ se ha asociado a la generación de radicales libres que causan peroxidación de lípidos y oxidación de proteínas entre otros daños. Se ha planteado que el Aβ pueda reconocer a receptores específicos que median a su vez neurotoxicidad. Entre estos se encuentra el receptor scavenger o pepenador que se expresa en la microglia y es capaz de internalizar agregados de este péptido. Independientemente de la vía de entrada del péptido a la célula, éste genera un estado de estrés oxidativo que eventualmente desencadena la muerte celular.
Estudios recientes desarrollados en nuestro laboratorio muestran que el proceso de traducción de proteínas que intervienen en el proceso de endocitosis mediada por un receptor puede ser afectado por una condición de estrés oxidativo. Este es el caso de la β–adaptina, proteína clave en la formación del pozo cubierto.
Palabras clave: Estrés oxidativo, péptido β–amiloide, microglia, receptor pepenador
Summary
Alzheimer's disease, the leading cause of dementia in the elderly is characterized by the presence in the brain of senile plaques formed of insoluble fibrillar deposits of β–amyloid peptide. This peptide is normally produced in a monomeric soluble form and it is present in low concentrations in the blood and spinal fluid. At physiological concentrations, this peptide is a neurotrophic and neuroprotector factor; nevertheless, with aging and particularly in Alzheimer's disease this peptide accumulates, favors the formation of insoluble fibrils and causes neurotoxicity.
β–amyloid peptide toxicity has been associated with the generation of free radicals that in turn promote lipid peroxidation and protein oxidation. Through the recognition of specific receptors such as the scavenger receptor, the β–amyloid peptide becomes internalizedin the form of aggregates. Independently of the way the peptide enters the cell, it generates oxidative stress that eventually triggers a state of neurotoxicity and cell death.
Recent studies in our laboratory have shown the effect caused by an extracellular oxidative stress upon the internalization of the scavenger receptor. We have also demonstrated that the process of protein translation of molecules implicated in the mechanism of endocytosis through the scavenger receptor, such as the case of β–adaptin, is arrested in microglial cells treated with β–amyloid.
Key words: Oxidative stress, β–amyloid peptide, microglial cells, scavenger receptor
Introducción
La enfermedad de Alzheimer es una de las causas más comunes de demencia en la población de edad avanzada. Su principal característica clínica es el deterioro de la capacidad cognoscitiva, que se da como consecuencia de la pérdida masiva y progresiva de neuronas en diferentes regiones del cerebro. El estudio histopatológico postmortem de cerebros de pacientes afectados con esta enfermedad muestra dos características morfológicas principales: marañas neurofibrilares y placas seniles o placas neuríticas;1 estructuras descritas por primera vez en 1907 por Alois Alzheimer.
Las marañas neurofibrilares son depósitos proteicos intracelulares formados por la proteína x estabilizadora asociada a microtúbulos (MAP–τ) que se encuentra hiperfosforilada, así como por neurofilamentos de mediano y alto peso molecular.
Esta MAP–τ modificada es resistente a cortes proteolíticos, lo cual sugiere que además de la fosforilación puede sufrir otras modificaciones como la glucosilación y la formación de puentes disulfuro que contribuyen a que se forme un entrecruzamiento entre monómeros de MAP–τ.2
Las placas seniles de la enfermedad de Alzheimer están formadas por un centro proteico rodeado de neuronas degeneradas,3 así como de células de glia y microglia.
El péptido β–amiloide (Aβ en su forma insoluble de depósitos fibrilares, es el principal componente proteico de estas placas. Esta observación realizada a mitad de los años 80's,4 llevó a pensar que los depósitos de este péptido pudieran corresponder al primer paso en la patogénesis de esta enfermedad y se les relacionó directamente con neurodegeneración.5 Esta hipótesis ganó terreno al demostrarse que el Aβ es tóxico para neuronas en cultivo.6,7
El péptido β–amiloide (Aβ en su forma insoluble de depósitos fibrilares, es el principal componente proteico de estas placas. Esta observación realizada a mitad de los años 80's,4 llevó a pensar que los depósitos de este péptido pudieran corresponder al primer paso en la patogénesis de esta enfermedad y se les relacionó directamente con neurodegeneración.5 Esta hipótesis ganó terreno al demostrarse que el Aβ es tóxico para neuronas en cultivo.6,7
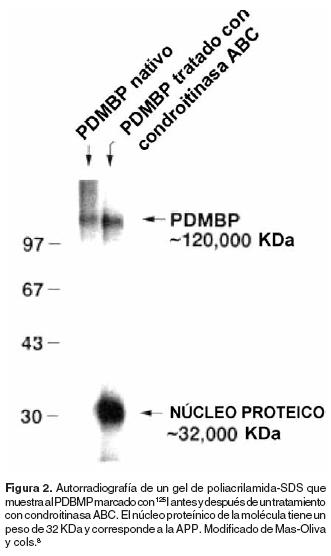
El Aβ puede tener entre 39 y 43 aminoácidos, presenta un peso molecular de 4 KDa y se deriva de un precursor de mayor tamaño denominado proteína precursora del péptido Aβ (APP)4 figura 1. La APP es una glicoproteína transmembranal que se deriva a su vez de un proteoglicano de mayor tamaño denominado PDMBP (Platelet–Derived Macrophage–Binding Proteoglycan) secretado en grandes cantidades por las plaquetas8 y que inhibe la endocitosis de LDL–Ac (Lipoproteínas de baja densidad acetiladas) en macrófagos.9 Existen tres isoformas de la APP: las isoformas de 751 y 770 aminoácidos encontradas tanto en neuronas como en células no neuronales, y la isoforma de 695 aminoácidos que se expresa en grandes cantidades en neuronas, (Figura 2).10
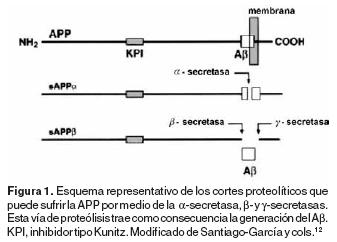
La APP puede sufrir un proceso proteolítico por la β–secretasa, que rompe al dominio que corresponde al péptido Aβ y que produce un precursor soluble (sAPP) con un peso molecular de entre 90–100 KDa.11
Además de las neuronas, las plaquetas también secretan sAPP, el cual compite con las LDL–Ac por la unión al receptor scavenger o pepenador (RS) clase A.12 Un procesamiento alternativo para APP llevado a cabo por la β– y γ–secretasas, rompe a la proteína precursora en los dominios extracelular y transmembranal, lo que la convierte en el péptido β–amiloide soluble (sAβ).13
Además de las neuronas, las plaquetas también secretan sAPP, el cual compite con las LDL–Ac por la unión al receptor scavenger o pepenador (RS) clase A.12 Un procesamiento alternativo para APP llevado a cabo por la β– y γ–secretasas, rompe a la proteína precursora en los dominios extracelular y transmembranal, lo que la convierte en el péptido β–amiloide soluble (sAβ).13
De la evidencia que sustenta la asociación entre APP y la patología del Alzheimer destaca lo siguiente:
1. Diferentes mutaciones de APP tienen relación con un aumento en la producción de Aβ.14,15
2. El gen de APP se localiza en el cromosoma 21, y en cerebros de individuos con trisomía 21 existen depósitos de Aβ16 lo que lleva eventualmente al desarrollo de esta enfermedad.17
3. Ratones que sobre–expresan APP presentan algunas características de la patología del Alzheimer.18
4. Mutaciones en las presenilinas, proteínas que regulan los cortes proteolíticos de APP, pueden contribuir a la patogénesis del Alzheimer al incrementar la producción de Aβ.19,20
Es importante aclarar que independientemente de que el Aβ juega un importante papel en el desarrollo de la enfermedad de Alzheimer, este padecimiento tiene varios componentes patofisiológicos que aún no se conocen con exactitud y que complican el entendimiento de la enfermedad.
Se han realizado estudios sobre la relación entre el número de placas seniles y "marañas" neurofibrilares con el grado de demencia de los pacientes con Alzheimer, encontrando que el número de "marañas" neurofibrilares y/o la extensión de la pérdida sináptica en la corteza cerebral está más relacionado con el grado de demencia que con el número de depósitos del péptido.21
Incluso las placas seniles pueden sorprendentemente estar en áreas del cerebro libres de anormalidades neuronales22 y en contraparte, la pérdida sináptica puede ocurrir en zonas completamente carentes de placas.23
El término "amiloide" hace referencia al almidón o a un compuesto parecido a la celulosa. Fue en 1851 cuando Virchow24 lo usó por primera vez para referirse a depósitos de polisacáridos que se tiñen con el colorante rojo Congo.25 Por otra parte, "amiloidosis" es un término usado en la medicina para describir cúmulos de péptidos que se presentan en varias enfermedades y en diferentes tejidos y que tienen la capacidad de agregarse como fibras insolubles. Estas fibras amiloidóticas forman filamentos no ramificados de entre 6 y 10 nm de diámetro y presentan una estructura secundaria de lámina β plegada, conformación que por lo general es indispensable para que estos péptidos sean tóxicos.26
El Aβ es producido normalmente en su forma monomérica soluble y circula en concentraciones bajas en el líquido cefalorraquídeo y sangre. Con el envejecimiento y sobre todo en la enfermedad de Alzheimer se forman agregados fibrilares insolubles. Existe una fuerte relación entre este proceso de formación de fibrillas y la capacidad del péptido para causar toxicidad en neuronas y activación de células de microglia.27
Papel fisiológico del péptido β–amiloide
A pesar del papel aparentemente negativo que presenta el Aβ en la enfermedad de Alzheimer, éste péptido participa, de una forma que no ha sido explicada con exactitud en varias funciones celulares normales, por ejemplo:
1. Ejerce una función autócrina y estimula la proliferación celular.28
2. Promueve la adhesión celular29 y protege a las neuronas contra daño oxidativo.30
3. El péptido sAβ puede interferir en procesos de señalización, vía proteínas G31 e incrementar la actividad de la MAP cinasa.32
4. En concentraciones fisiológicas puede actuar como factor neurotrófico y neuroprotector.33,34
5. Se ha planteado que es un regulador fisiológico de la función de canales iónicos (K+, Ca2+) en neuronas35,36 y que es secretado por algunas de éstas células en respuesta a la actividad neuronal para regular negativamente la transmisión sináptica excitatoria.37
6. Se ha propuesto que los depósitos de Aβ puedan atrapar iones metálicos potencialmente peligrosos.38,39
7. Concentraciones nanomolares del péptido pueden bloquear la apoptosis neuronal provocada por la ausencia de factores tróficos.40
El Aβ posee una alta afinidad por Cu2+ y Zn2+ y también puede catalizar la dismutación del radical superóxido (O2) a H2O2, actuando entonces como antioxidante.41,42
Se han detectado depósitos de Aβ en cerebros humanos después de daño traumático,43 y se sabe que tanto el péptido 1–40, pero especialmente el 1–42, aumentan su concentración en el fluido cerebroespinal durante la primera semana que sigue a un traumatismo encefálico.44
Se han detectado depósitos de Aβ en cerebros humanos después de daño traumático,43 y se sabe que tanto el péptido 1–40, pero especialmente el 1–42, aumentan su concentración en el fluido cerebroespinal durante la primera semana que sigue a un traumatismo encefálico.44
Algunos grupos de estudio han mostrado que el número de placas de Aβ y de marañas neurofibrilares en la corteza cerebral están correlacionadas inversamente con el estrés oxidativo,45–48 por lo que cuando la concentración del Aβ se incrementa, el daño citoplásmico disminuye. En pacientes con síndrome de Down también existe correlación negativa entre los depósitos de Ap y daño oxidativo.49
Sin embargo, a pesar de que se han aislado oligómeros solubles del Ap tanto de cerebros normales como de cerebros afectados por la enfermedad de Alzheimer;50 los niveles del péptido soluble suelen ser mayores en pacientes con esta enfermedad que en sujetos normales, siendo mayor la proporción del péptido 1–42 que la del péptido 1–40.51
El péptido 1–42 es menos soluble que las otras isoformas del péptido, desarrolla fibrillas mucho más rápido52 y promueve la agregación de formas más pequeñas como la 1–40.
El péptido 1–42 es menos soluble que las otras isoformas del péptido, desarrolla fibrillas mucho más rápido52 y promueve la agregación de formas más pequeñas como la 1–40.
Por otra parte, mediante la utilización de ensayos in vitro se sabe que los péptidos que contienen al fragmento hidrofóbico 29–35 (GAIIGLM) forman lentamente agregados estables y se transforman en neurotóxicos después de un proceso de incubación. En cambio el fragmento 25–35 (GSNKGAIIGLM), descrito como la parte más tóxica del péptido, se agrega rápidamente y es neurotóxico de forma inmediata.53 Todos estos estudios han demostrado que es necesaria la transición del péptido de una forma soluble monomérica a una insoluble o fibrilar para que sean tóxicos a la célula y que esto es el resultado de un cambio al azar en su estructura secundaria (random coil) y α–hélice a una de lámina p.54,55 Asimismo, en 1999 Walsh y colaboradores56 reportaron que los monómeros y dímeros del Aβ no son tóxicos para las células, mientras que oligómeros agregados de bajo peso molecular llamados protofibrillas sí lo son.
Iones metálicos y su interacción con el péptido β–amiloide
Diversos estudios indican que la presencia de iones metálicos puede ser fundamental tanto para la fibrilización del Aβ, como para la iniciación de la generación de especies de oxígeno reactivas (ERO) relacionadas con el proceso de estrés oxidativo.57,58 Mediante ensayos in vitro se conoce que en ausencia de estos iones metálicos, el Aβ es monomérico, presenta una conformación a–hélice y no forma agregados.59
Estudios epidemiológicos han sugerido una alta incidencia de la enfermedad de Alzheimer asociada con niveles ambientales elevados de Al3+, Zn2+ y Fe3+.60 Asimismo, se ha detectado un alto contenido de Al3+, Fe3+, Cu2+ y Zn2+ en el cerebro de pacientes con esta enfermedad.61 La concentración de estos iones metálicos puede estar incrementada entre 3 y 5 veces al compararse con la de cerebros de pacientes de la misma edad que no presentan esta patología.62–65 Se han detectado iones metálicos en el centro y la periferia de las placas seniles,66 los que pueden ser tóxicos no sólo por favorecer la generación de ERO sino también estimulando directamente la formación de fibrillas.38 Mediante el uso de ensayos in vitro, se ha detectado que concentraciones traza de Fe3+, Al3+ y Zn2+ estimulan la agregación del Ap entre 100 y 1000 veces.67
Una amplia gama de sistemas generadores de radicales libres de oxígeno, enzimáticos o no enzimáticos, son capaces de catalizar modificaciones oxidativas de proteínas cuando el Fe3+ o el Cu2+ están en presencia de O2 y de un donador electrónico apropiado.68 La conversión de radicales O2– y H2O2 al radical hidroxilo (HO*) puede llevarse a cabo sólo cuando están presentes metales de transición en concentraciones catalíticas.69
Bush y colaboradores en 199941 sugirieron que el Cu2+ juega un papel importante en el estrés oxidativo producido por el Aβ. Estos investigadores proponen que el Ap provee de un electrón al Cu2+ unido al péptido, formando Cu1+. Sin embargo, el Ap 1–40, que tiene menor afinidad por el Cu2+ y el 25–35 que no presenta afinidad por este ión, también son tóxicos.7,70
Atwood y colaboradores,38,42 han caracterizado al Aβ como una metaloproteína que une iones de elementos de transición por medio de tres histidinas (posiciones 6,13 y 14) y una tirosina (posición 10), localizadas en la parte hidrofílica N–terminal del péptido, además de la metionina 35 en la región lipofílica C–terminal.71–73 Algunos estudios indican que los residuos de histidina son importantes para la agregación del Ap, ya que la ausencia de histidinas,74 o la modificación de estos residuos, disminuye notablemente la agregación del Ap promovida por el Cu2+, Zn2+ o Fe3+.38 Estos hallazgos pueden explicar la razón por la cual no se detectan depósitos de Ap en el cerebro de la rata, pues en ésta especie el Aβ carece de histidinas.74
Por otra parte, los péptidos que pierden la metionina 35 tienen una baja capacidad de reducir al Cu2+. La sustitución de este residuo por otro aminoácido elimina la acción prooxidante del péptido 25–3575 y disminuye la oxidación de proteínas, así como su neurotoxicidad.76 Sin embargo, la presencia de la metionina 35 no es suficiente para explicar la actividad redox del péptido y su neurotoxicidad, ya que en la rata el Aβ que posee la metionina 35 carece del sitio clave de unión a iones metálicos (histidina 13).
El grupo de Butterfield ha propuesto un modelo de estrés oxidativo para explicar la neurotoxicidad del Ap basada en el residuo metionina 35. Este aminoácido está asociado con la formación de fibrillas y la generación de estrés oxidativo, lo cual se ha demostrado en ensayos in vitro en los que se ha sustituido este residuo por norleucina, un aminoácido del mismo tamaño y la misma hidrofobicidad que la metionina, pero con un grupo –CH2 en lugar del átomo de azufre del grupo tioeter de la metionina. Este cambio no provoca oxidación de proteínas o toxicidad en ensayos realizados con neuronas de hipocampo aún en presencia de Cu2+.77
La metionina 35 es el residuo más susceptible a la oxidación in vivo, especialmente bajo condiciones de estrés oxidativo.78 El estudio de placas seniles que contienen al Aβ 1–40 muestra una gran proporción de metionina sulfóxido,79 y se conoce, por estudios de modelaje estructural, que la oxidación de residuos de metionina altera significativamente la estructura secundaria de los mismos.80 Particularmente la oxidación de la metionina induce una estructura de lámina p.10 La razón por la que la metionina 35 confiere propiedades neurotóxicas al Aβ es desconocida, sin embargo existe evidencia de que al parecer se necesitan trazas de iones metálicos para que esto suceda.
Radicales libres y estrés oxidativo
Un radical libre es un átomo o molécula, neutra o cargada, que contiene uno o más electrones desapareados, ya sea por pérdida o ganancia de estos. La existencia de electrones desapareados incrementa la reactividad de la especie química, pues estos electrones tienden a aparearse con un electrón libre de otro átomo o molécula. La producción de radicales libres ocurre por la adición o remoción de un electrón en una reacción de óxido–reducción y se producen normalmente durante el metabolismo de la célula.81
A pesar del daño que pueden producir los radicales libres, debe mencionarse que tienen funciones normales dentro del metabolismo celular tales como transducción de señales y expresión génica, activación de factores transcripcionales y protección inmunológica.82
El oxígeno (O2) es un elemento esencial para la vida, pero puede llegar a ser tóxico al formar especies reactivas de oxígeno (ERO). Estas son producto de la ruptura o de la excitación del O2, como son el oxígeno atómico (O), el ozono (O3) y el oxígeno en singulete (1O2). Otras están parcialmente reducidas como lo es el peróxido de hidrógeno (H2O2), el radical superóxido (O2) y el radical hidroxilo (HO*).83 El 1O2 es muy reactivo y puede reaccionar con la mayoría de moléculas celulares.84 El O2 y el H2O2 son poco reactivos, sin embargo ambos generan 1O2 y HO*, compuestos altamente tóxicos. Cuando el H2O2 acepta un electrón desapareado, que puede provenir de un metal de transición como el Fe2+ o el Cu1+, forma HO* y HO (reacción de Fenton) (Fe2+ + H2O2 –––> Fe3+ + HO• + HO ).
La toxicidad del O2 y del H2O2 depende en gran medida de la disponibilidad y la distribución de estos metales de transición.85 Además de estas especies reactivas de oxígeno, pueden formarse otras de importancia biológica que se combinan con nitrógeno (ERN), como el monóxido de nitrógeno u óxido nítrico (NO•).86
En los organismos aerobios, la producción de ERO y ERN se encuentra en equilibrio con los sistemas de defensa antioxidantes. Sin embargo, al romperse este equilibrio las especies reactivas aumentan, se producen daños al ADN, a proteínas y a lípidos, lo que se conoce como estrés oxidativo. Estos daños pueden ser reversibles al ser reparados o al reemplazar a las moléculas dañadas. Sin embargo, si en la célula se producen especies reactivas en exceso, o bien, los sistemas antioxidantes de la célula no responden adecuadamente, la célula puede morir.83
Existen varios sistemas de protección en la célula que evitan el excesivo incremento de especies reactivas, entre los que se encuentran: la actividad de la superóxido dismutasa (SOD), la catalasa y la glutation peroxidasa (GSH peroxidasa). La superóxido dismutasa es una enzima mitocondrial que convierte al O2 en H2O2. La glutation peroxidasa y la glutation reductasa trabajan en conjunto, siendo la GSH peroxidasa una enzima citosólica que transforma al H2O2 en dos moléculas de glutation reducido (GSH) que ceden dos hidrógenos, lo cual promueve que se forme entre ellas un enlace disulfuro (GSSG). El glutation se regenera rápidamente mediante la glutation reductasa en presencia de NADPH. Finalmente la catalasa, enzima confinada a los peroxisomas, destruye al H2O2 por dismutación. Existen además otros sistemas de protección que eliminan a los radicales libres como el ácido úrico, la bilirrubina y la albúmina, cuya acción es inespecífica y poco eficiente. Asimismo, las vitaminas C y E así como los flavonoides son capaces de inactivar a los radicales libres.82
Estrés oxidativo y enfermedad Alzheimer
El cerebro es particularmente vulnerable al estrés oxidativo ya que presenta una elevada tasa metabólica derivada de la glucosa, posee niveles muy bajos de defensas antioxidantes, contiene altas concentraciones de ácidos grasos poliinsaturados, que son posible blanco de peroxidación lipídica, y además es rico en actividades enzimáticas relacionadas con metales de transición, los cuales pueden catalizar la formación de radicales libres.87
Aunque el mecanismo por el cual el Aβ genera radicales libres en la enfermedad de Alzheimer no se conoce, se ha definido que estas especies reactivas causan peroxidación de lípidos, oxidación de proteínas y pérdida de integridad de la membrana. Esto trae como consecuencia la inhibición de ATPasas, pérdida de la homeostasis del calcio, inhibición del sistema de captura de glutamato dependiente de sodio en células gliales, alteración de vías de señalización, activación de factores transcripcionales y finalmente apoptosis.88
Las evidencias que indican que la citotoxicidad del Aβ es mediada a través de radicales libres son variadas:
1) Concentraciones micromolares de Aβ provocan un aumento en los niveles de H2O2 en células en cultivo, sin embargo la presencia de catalasa o de SOD89 previene la toxicidad del péptido.
2) Células seleccionadas por su resistencia al Aβ son también altamente resistentes al efecto del H2O2 y contienen altos niveles del glutation.90
3) Por otro lado, se han detectado niveles altos de ERO intracelulares en individuos con síndrome de Down que sobreexpresan al gen de la APP.91
Aunque por mucho tiempo se ha pensado que el generador de ERO en la enfermedad de Alzheimer es exclusivamente el Aβ, se ha planteado también la existencia de otras fuentes de radicales libres, tales como el Fe3+, que se encuentra incrementado en las marañas neurofibrilares y en los depósitos de Aβ.92 El Fe3+, cataliza la formación de HO• a partir de H2O2, así como la formación de productos de glucosilación avanzada o AGE (Advanced Glycation End Products). Al3+por su parte, también se acumula en las marañas neurofibrilares,63 estimulando la peroxidación lipídica inducida por Fe3+.93
Por otra parte, la microglia activada que rodea las placas seniles94 es una fuente de NO y O2,95 los que a su vez pueden reaccionar para formar peroxinitrito (ONOO–).96 Los AGE en presencia de metales de transición pueden también producir especies reactivas.97 Además, tanto los AGE como el Aβ pueden reconocer receptores demembrana específicos tales como RAGE (Receptor– Advanced Glycation End products) y RS (Receptor Scavenger), mecanismo que a su vez puede incrementar la producción de ERO.98
A pesar de las controversias al respecto, es un hecho que el cerebro de pacientes con enfermedad de Alzheimer presenta un estado de estrés oxidativo severo, detectándose un aumento en la peroxidación de lípidos, bajos niveles de ácidos grasos poliinsaturados (PUFA, Polyunsatured Fatty Acids) y un incremento en el 4–hidroxinonenal (HNE), un producto aldehídico neurotóxico proveniente de la oxidación de los PUFA. El HNE puede difundir desde el sitio de su producción, modificar organelos neuronales y alterar su función.99 El daño oxidativo que se presenta en el Alzheimer incluye AGE,100 productos de nitrosilación,96 proteínas de neurofilamentos carbonil–modificadas y carbonilos libres.101
Asimismo, existe un incremento en la oxidación proteica y un aumento en la oxidación del ADN detectado por niveles altos de su producto de degradación, el 8–hidroxi–2'–desoxiguanosina (8–OHdG). El aumento de este último compuesto, sin embargo, no siempre se lleva a cabo, pues se han encontrado niveles muy bajos de 8–OHdG en algunos casos de Alzheimer con grandes depósitos de Aβ.102 Estos hallazgos pueden parecen contradictorios, pero dan evidencia de que el Aβ y las marañas neurofibrilares, pueden constituir una respuesta de la célula al estrés oxidativo al tener funciones anti–oxidantes, al menos en las etapas iniciales de la enfermedad.102
Con respecto a la respuesta inmune, se sabe que en procesos normales de inflamación se generan ERO que dañan al tejido circundante. A pesar de que la inflamación aguda, que incluye edema e invasión de neutrófilos no es característica de la enfermedad de Alzheimer,103 existe evidencia de daño celular mediado por células del sistema inmune. Una evidencia que sustenta la posibilidad de un proceso inflamatorio en el cerebro de pacientes con enfermedad de Alzheimer, es el hecho de que una terapia basada en drogas anti–inflamatorias no esteroideas trae beneficios cognitivos.104,105 Una fuente potencial importante de ERO en el cerebro es la microglia,106 que de forma similar a los monocitos de la sangre o a los macrófagos peritoneales, son células que una vez activadas presentan una actividad fagocítica alta.107 La microglia activada in vitro produce O2~, lo cual sugiere que el Aβ es indirectamente neurotóxico por la activación de la microglia y la consecuente generación de radicales libres.108
La respuesta inflamatoria secundaria a la acción del Aβ fue estudiada primeramente en macrófagos peritoneales.109 En estas células, el Aβ 25–35 y el 1–40 son capaces de estimular a la NADPH oxidasa, una fuente importante de O2–.110 El péptido 1–40 causa además un incremento en la producción de NO. Asimismo, se ha detectado la formación de NO en microglia111 por un aumento en la actividad de la óxido nítrico sintasa causada por el péptido. El NO resultante puede reaccionar rápidamente con el radical O2– que se produce en la mitocondria y formar ONOO–. Además, la exposición de microglia al Aβ provoca un aumento en la expresión de varias interleucinas como TNF–α y TGF–β entre otras sustancias moduladoras de las cascadas inmunes.112
Por otra parte, se sabe que el metabolismo energético de las neuronas está deteriorado en cerebros afectados con la enfermedad de Alzheimer. La tomografía de emisión de positrones de pacientes con Alzheimer muestra una disminución progresiva en el metabolismo de la glucosa en el cerebro y un menor flujo sanguíneo en los lóbulos parietal y temporal, que correlaciona con el grado de demencia de los pacientes.113 La primera evidencia de una posible relación entre el estrés oxidativo y alteraciones en el transporte de glucosa fue encontrada por estudios de neuronas en cultivo, en los que se encontró que el Aβ altera la captura de glucosa, lo que causa a su vez un decremento en los niveles de ATP. Se ha sugerido que esto es provocado por la conjugación HNE con la proteína transportadora de glucosa, GLUT3.114 Las alteraciones en el metabolismo de la glucosa en el cerebro limitan la síntesis de acetil–colina, glutamato, aspartato, ácido–y–aminobutírico (GABA), glicina y ATP.115
Péptido β–amiloide y sus receptores
Se ha propuesto la existencia de receptores membranales específicos que medían la citotoxicidad inducida por el Aβ,116 entre ellos RAGE.117,118 Este es un receptor de superficie celular relacionado con la producción intracelular de ERO, que puede inducir la migración de microglia hacia depósitos de Aβ.97,98 La proteína x asociada a microtúbulos, es por ejemplo, blanco de la formación de AGE intracelulares. Sin embargo, también existe evidencia de que la toxicidad del Aβ puede ser independiente de este receptor, ya que no se ha encontrado al ARNm del RAGE en neuronas corticales o células PC12.119
Otro de los receptores que ha sido implicado en la toxicidad del Aβ es el receptor nombrado LRP (LDL Related Receptor Protein), que une diversos ligandos incluyendo quilomicrones, apolipoproteína E y α2–macroglobulina.120 Ésta última es una proteína cuya unión al Aβ parece favorecer su internalización.121 LRP puede unir e internalizar a APP soluble, así como una isoforma no procesada amiloidogénica, la cual a su vez puede llevar a la formación de Aβ.122 Evidencia reciente indica que LRP de células de microglia puede jugar un papel importante como un receptor del Aβ123
Otro candidato es la glicoproteína 330 (gp330/megalina), un receptor de membrana que pertenece a la familia de los receptores de LDL y que tiene múltiples ligandos entre los que se incluye a la apoJ. Este receptor ha sido implicado en el transporte a través de la barrera hematoencefálica del Aβ formando un complejo con la apoJ.124
Finalmente, existe evidencia de que el RS está implicado en la internalización y toxicidad del Aβ. El RS se expresa en células de glia y microglia y es capaz de internalizar al Aβ en forma de agregados.125,126 Evidencia reciente muestra que el RS puede iniciar el daño mediado por inmunidad en la enfermedad de Alzheimer, hecho que es avalado por la fuerte expresión de este receptor asociado a la presencia de placas seniles.127
Receptores "scavenger"
El RS constituye parte de una amplia familia de receptores de membrana que se caracterizan por su habilidad para internalizar LDL acetiladas y LDL oxidadas,128 además de una gran variedad de ligandos, muchos de ellos caracterizados por sus propiedades polianiónicas.129 Uno de los ligandos recientemente descritos para el RS–A es la molécula paramiosina, una proteína proveniente de Taenia soleum, que parece favorecer la formación de arreglos tridimensionales del propio receptor, los que facilitan la formación del pozo cubierto y la endocitosis del mismo.130
El contenido de colesterol en la membrana plasmática, se ha observado también influye en la formación de estos arreglos tridimensionales entre los receptores RS–A,131 más aún, este arreglo tridimensional del RS–A favorece la síntesis de proteínas accesorias en el proceso de endocitosis.130
El contenido de colesterol en la membrana plasmática, se ha observado también influye en la formación de estos arreglos tridimensionales entre los receptores RS–A,131 más aún, este arreglo tridimensional del RS–A favorece la síntesis de proteínas accesorias en el proceso de endocitosis.130
El RS está implicado en la generación de células espumosas derivadas de macrófagos y de la consecuente formación de la placa aterosclerótica en el sistema circulatorio. De igual forma, participa en funciones de inmunidad innata, eliminación de células apoptóticas, defensa del hospedero, transporte reverso del colesterol y adhesión celular. Este receptor se encuentra localizado a concentraciones importantes en macrófagos y células afines como la microglia, así como en hepatocitos, ovario, testículo, glándulas suprarrenales y astrocitos.132
Un posible papel del RS–A en la inducción de la enfermedad de Alzheimer ha sido sugerido por datos que muestran que este receptor puede promover la adhesión de fibrillas de Aβ a células de microglia,133 además de poder mediar su endocitosis125 y su degradación.134,135 Asimismo, la expresión del RS–A está aumentada en la microglia de cerebros de pacientes con EA y en el cerebro de ratones transgénicos que expresan una forma mutada de la proteína precursora del A(3 (APP23) y los cuales desarrollan una patología parecida a la de la enfermedad de Alzheimer.136 El RS–BI localizado en microglia neonatal también parece ser parte de la unión del A(3 a estas células y de la producción de ERO,137,138 además de participar en la endocitosis del péptido.139,140
La interacción in vitro de fibras de Aβ con microglia neonatal estimula a estas células a producir sustancias proinflamatorias y potencialmente neurotóxicas como lo son el óxido nítrico, el factor de necrosis tumoral α (TNF–α), ERO, ERN, etc.126,141 Se ha encontrado que la remoción de células de microglia de cultivos mixtos de células nerviosas y fibras amiloides elimina casi totalmente el efecto tóxico de las fibrillas sobre las neuronas142 sugiriendo que la microglia, y/ o moléculas que éstas producen median los efectos neurotóxicos de las fibrillas del Aβ.
El trabajo realizado en nuestro laboratorio muestra que la capacidad de internalización del RS–A disminuye en condiciones de estrés oxidativo extracelular.143 La unión del ligando (LDL–Ac), la expresión del RNAm–RS–A y la misma estructura del RS–A no se modifican en condiciones de estrés oxidativo extracelular, y sin embargo, el RS–A no puede ser endocitado.143 Por lo tanto, ¿cuál es el mecanismo afectado que impide la internalización del receptor secundario a la presencia de estrés oxidativo? Nuestros estudios utilizando al Aβ como ligando del RS e inductor de estrés oxidativo fisiológico demuestran que una de las múltiples proteínas implicadas en el proceso de endocitosis mediada por clatrina (clatrina, α– y β–adaptina, CALM, eps15), en específico la β–adaptina disminuye de forma importante en células de microglia expuestas a este péptido (Figura 3). Esta disminución asociada a una alteración en la traducción de la proteína coincide con la incapacidad de la célula para endocitar al Aβ. La pérdida en el nivel de concentración crítica de esta proteína, clave en la formación del pozo cubierto y por tanto en la endocitosis, podría ayudar a explicar la acumulación de agregados de Aβ extracelular que se presentan en la enfermedad de Alzheimer.144
Tomando en cuenta que la enfermedad de Alzheimer es un desorden neurodegenerativo complejo cuya patogénesis no se conoce con exactitud, los hallazgos bioquímicos y genéticos que se conocen hasta el momento solamente permiten entender parcialmente la muerte neuronal, por lo que continuar con el estudio de los factores tanto extra como intracelulares que controlan el proceso, sin duda mejorará el conocimiento de la fisiopatología de este padecimiento. De estos factores, el estrés oxidativo localizado y en especial su repercusión sobre la traducción de proteínas que participan en la formación del pozo cubierto, parece constituir un nuevo elemento que deberá ser estudiado a profundidad en el afán de entender mejor y poder algún día llegar a contrarrestar el desarrollo de la enfermedad.
Agradecimientos
Las investigaciones realizadas por los autores recibieron el apoyo de CONACYT y la DGAPA–UNAM, México. Agradecemos a la Maestra Blanca Delgado–Coello su apoyo técnico y a la Sra. Ma. Elena Gutiérrez su apoyo secretarial.
Referencias
1. Terry RD, Masliah E, Salmon DP, Butters N, De Teresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease : synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991; 30:572–580. [ Links ]
2. Durany N, Munch G, Michel T. Investigations on oxidative stress and therapeutical implications in dementia. Eur Arch Psychiatry Clin Neurosci 1999; 249:S68–S73 [ Links ]
3. Katzman R, Saitoh T. Advances in Alzheimer's disease. FASEB J 1991; 4:278–286. [ Links ]
4. Glenner G, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 1984; 120:885–890. [ Links ]
5. Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 1985;82:4245–4249. [ Links ]
6. Pike CJ, Walenzcewicz AJ, Glabe CG, Cotman CW. In vitro aging of β–amyloid protein causes peptide aggregation and neurotoxicity. Brain Res 1991; 563:311–314. [ Links ]
7. Varadarajan S, Yatin S, Kanski J, Jahanshahi F, Butterfield DA. Methionine residue 35 is important in amyloid β–peptide–associated free radical oxidative stress. Brain Res Bull 1999; 50:133–141. [ Links ]
8. Mas–Oliva J, Arnold KS, Wagner WD, Phillips DR, Pitas RE, Innerarity TL. Isolation and characterization of a Platelet–derived Macrophage–binding proteoglycan. J Biol Chem 1994; 269:10177–10183. [ Links ]
9. Mas–Oliva J, Arnold KS, Wagner WD, Innerarity TL. Isolation of a platelet proteoglycan that inhibits the uptake of acetyl LDL by macrophages. Circulation 1992; 86:I–156. [ Links ]
10. Selkoe DJ. Cell biology of the amyloid p–protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol 1994; 10:373–403. [ Links ]
11. Sisodia SS. p–amyloid precursor protein clavage by a membrane–bound protease. Proc Natl Acad Sci USA 1992; 89:6075–6079. [ Links ]
12. Santiago–Garcia J, Mas–Oliva J, Innerarity TI, Pitas RE. Secreted forms of the amyloid–β–precursor protein are ligands for the A scavenger receptor. J Biol Chem 2001; 276:30655–30661. [ Links ]
13. Busciglio J, Gabuzda DH, Matsudairia P, Yankner BA. Generation of p–amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA 1993; 90:2092–2096. [ Links ]
14. Selkoe DJ. Amyloid p–protein and the genetics of Alzheimer's disease. J Biol Chem 1996; 271:18295–18298. [ Links ]
15. Sandbrick R, Hartmann T, Masters CL, Beyreuther K. Genes contibuting to Alzheimer's disease. Mol Psychiat 1996, 1:27–40. [ Links ]
16. Teller JK, Russo C, DeBusk LM, Angelini G, Zaccheo D, Dagna–Bricarelli F, et al. Presence of soluble amyloid β–peptide precedes amyloid plaque formation in Down's syndrome. Nat Med 1996; 2:93–95. [ Links ]
17. Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann Neurol 1985; 17:278–282. [ Links ]
18. Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL et al. Amyloid β deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 1998; 70:2212–2215. [ Links ]
19. Hardy J. Amyloid, the presenilinas and Alzheimer's disease. Trends Neurosci 1997; 20:154–159. [ Links ]
20. Selkoe DJ. Alzheimer's disease: genotypes, phenotypes, and treatments. Science 1997; 275:630–631. [ Links ]
21. Morris JC, McKeel DW Jr, Storandt M, Rubin EH, Price JL, Grant EA, et al. Very mild Alzheimer's disease: informant–based clinical, psychometric and pathologic distinction from normal aging. Neurology 1991 ;41:469–478. [ Links ]
22. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Balu AD, Davies P et al. Identification of normal and pathological aging in prospectively studied non–demented elderly humans. Neurobiol Aging 1991; 13:179–189. [ Links ]
23. Cataldo AM, Barnett JL, Berman SA, Li J, Quarless S, Bursztaijn S, et al. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up–regulation of the endosomal–lysosomal system. Neuron 1995; 14:671 –680. [ Links ]
24. Behl C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Progress in Neurobiol 1999; 57:301–323. [ Links ]
25. Smith MA, Rudnicka–Nawrot M, Richey PL, Praprotnik D, Mulvihill P, Miller CA, et al. Carbonyl–related posttranslational modifications of neurofilament protein in the neurofibrillary pathology of Alzheimer's disease. J Neurochem 1995; 64:2660–2666. [ Links ]
26. Xing Y, Higuchi K. Amyloid fibril proteins, Mech Ageing Dev 2002; 123:1625–1636. [ Links ]
27. Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron 1996; 16:921–932. [ Links ]
28. Saitoh T, Sundsmo M, Roch J, Ximura M, Cole G, Schubert D, et al. Secreted form of amyloid p protein precursor is involved in the growth regulation of fibroblasts. Cell 1989; 58:615–622. [ Links ]
29. Schubert D, Jin LW, Saitoh T, Cole G. The regulation of amyloid p protein precursor secretion and its modulatory role in cell adhesion. Neuron 1989; 3:689–694. [ Links ]
30. Goodman Y, Mattson MP. Secreted forms of p–amyloid precursor protein protect hippocampal neurons against amyloid β–peptide–induced oxidative injury. Exp Neurol 1994; 128:1–12. [ Links ]
31. Nishimoto I, Okamato T, Matsuura Y, Takahashi S, Oka Mato T, Murayama Y, et al. Alzheimer amyloid protein precursor complexes with brain GTP– binding protein Go. Nature 1993; 362:75–79. [ Links ]
32. Greenberg S, Koo E, Selkoe D, Qiu W, Kosik K, Secreted β–amyloid precursor protein stimulates MAP–kinase and enhances tau phosphorylation. Proc Natl Acad Sci USA 1994; 91:7104–7108. [ Links ]
33. Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid p protein: reversal by tachykinin neuropeptides. Science 1990; 250:279–282. [ Links ]
34. Postuma RB, He W, Nunan J, Beyreuther K, Masters CL, Barrow CJ, et al. Substrate–bound β–amyloid peptides inhibit cell adhesion and neurite outgrowth in primary neuronal cultures. J Neurochem 2000; 74:1122–1130. [ Links ]
35. Ramsden M, Plant LD, Webster NJ, Vaughan PF, Henderson Z, Pearson HA. Differential effects of unaggregated and aggregated amyloid β protein (1–40) on K+ channel currents in primery cultures of rat cerebellar granule and cortical neurons. J Neurochem 2001; 79:699–712. [ Links ]
36. Ramsden M, Henderson Z, Pearson HA. Modulation of Ca2+ channel currents in primary cultures of rat cortical neurons by amyloid p protein (1–40) is dependent on solubitity status. Brain Res 2002; 956:254–261. [ Links ]
37. Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron 2003; 37:925–937. [ Links ]
38. Atwood CS, Huang X, Moir RD, Bacarra NM, Romano D, Tanzi RE, et al. Dramatic aggregation of Alzheimer Aβ by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem 1998; 273:12821–1286. [ Links ]
39. Smith MA, Nunorama A, Zhu X, Takeda A, Perry G. Metabolic, metallic, and mitotic sources of oxidative stress in Alzheimer disease. Antioxid Redox Signal 2000; 12:13–20. [ Links ]
40. Chan CW, Dharmarajan A, Atwood CS, Huang X, Tanzi RE, Bush AI, Martins RN. Anti–apoptotic action of Alzheimer Aβ. Alzheimer's Reports 1999; 2:1–6. [ Links ]
41. Bush AI, Lynch T, Cherny RC, Atwood CS, Goldstein LE, Moir RD, et al. Alzheimer Aβ functions as a superoxide antioxidant in vitro and in vivo. Soc Neurosci Abstrc 1999; 25:14. [ Links ]
42. Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP et al. Characterization of copper interactions with Alzheimer amyloid p peptides: identification of an attomolar–affinity copper binding site on amyloid p 1–42. J Neurochem 2000; 75:1219–1233. [ Links ]
43. Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. –amyloid precursor protein as a marker for axonal injury after head injury. Neurosci Lett 1993; 160:139–144. [ Links ]
44. Raby CA, Morganti–Kossmann MC, Kossmann T, Stabel MD, Watson MD, Evans LM, et al. Traumatic brain injury increases β–amyloid peptide 1–42 74. in cerebrospinal fluid. J Neurochem 1998; 71:2505–2509. [ Links ]
45. Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, et al. Evidence that the β–amyloid plaques of Alzheimer's disease represent the redox–silencing and entombment of Ap by zinc. J Biol Chem 75. 2000; 275:1 9439–1 9442. [ Links ]
46. Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, et al. Oxidative damage is the earliest event in Alzheimer's disease. J Neuropathol Exp Neurol 2001; 60:759–767. [ Links ]
47. Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer's disease. Biochem Biophys Acta 2000; 1502:139–144. [ Links ]
48. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci USA 1994; 91:10771 –10778. [ Links ]
49. Nunomura A, Perry G, Pappolla RP, Friedland RP, Hirai K, Chiba S, et al. Neuronal oxidative stress precedes amyloid–β deposition in Down syndrome. J Neuropathol Exp Neurol 2000; 59:1011–1017. [ Links ]
50. Kuo YM, Emmerling MR, Vigo–Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, et al. Water–soluble Aβ (N–40, N–42) oligomers in normal and Alzheimer disease brains. J Biol Chem 1996; 271:4077–4081. [ Links ]
51. Funato H, Yoshimura M, Kusui K, Tamaoka A, Ishikawa K, Ohkoshi N, et al. Quantitation of amyloid β–protein (αβ) in the cortex during aging and in Alzheimer's disease. Am J Pathol 1998; 152:1633–1640. [ Links ]
52. Wisniewski T, Ghiso J, Frangione B. Biology of β–amyloid in Alzheimer's disease. Neurobiol Dis 1997; 4:313–328. [ Links ]
53. Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β–amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 1993; 13:1676–1687. [ Links ]
54. Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, et al. Secondary structure of amyloid β peptide correlates with neurotoxic activity in vitro. Mol Pharmacol 1994; 45:373–379. [ Links ]
55. Iversen LL, Mortishire–Smith RJ, Pollack SJ, Shearman MS. The toxicity in vitro of β–amyloid protein. Biochem J 1995; 311:1–16. [ Links ]
56. Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, et al. Amyloid–protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem 1999; 274:25945–25952. [ Links ]
57. Gerlach M, Ben–Shachar D, Riederer P, Youdim MB. Altered brain metabolism of iron as a cause of degenerative disease? J Neurochem 1994; 63:793–807. [ Links ]
58. Bondy SC, Guo–Ross SX, Truong AT. Promotion of transition metal–induced reactive oxygen species formation by β–amyloid. Brain Res 1998; 799:91–96. [ Links ]
59. Liu ST, Howlett G, Barrow CJ. Histidine–13 is a crucial residue in the zinc ion–induced aggregation of the Aβ peptide of Alzheimer's disease. Biochemistry 1999; 38:9373–9378. [ Links ]
60. Manthy PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, Maggio JE. Aluminum, iron and zinc ions promote aggregation of physiological concentrations of β–amyloid peptide. J Neurochem 1993; 61:1171–1174. [ Links ]
61. Good PF, Perl DP, Bierer LM, Schmeidler J. Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer's disease: a laser microprobe (LAMMA) study. Ann Neurol 1992; 31:286–292. [ Links ]
62. Richardson JS. Free radicals in the genesis of Alzheimer's disease. Ann NY Acad Sci. 1993; 695:73–76. [ Links ]
63. Basun H, Forssell LG, Wetterberg L, Winblad, B. Metals and trace elements in plasma and cerebrospinal fluid in normal aging and Alzheimer's disease. J Neural Transm 3, 1991; 4:231–258. [ Links ]
64. Castellani RJ, Smith MA, Nunomura A, Harris PL, Perry G. Is increased redox–active iron in Alzheimer disease a failure of the copper–binding protein cerulosplasmin? Free Radic Biol Med 1999; 26:1508–512. [ Links ]
65. Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron regulatory proteins in the brain in aging and Alzheimer's disease. J Neurosci Res 1992; 31:327–335. [ Links ]
66. Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc Alzheimer's disease senile plaques. J Neurol Sci 1998; 158:47–452. [ Links ]
67. Dyrks T, Dyrsk E, Hartmann T, Masters C, Beyreuter K . Amyloidogenicity of βA4 and βA4–bearing amyloid protein precursor fragments by metal–catalyzed oxidation. J Biol Chem 1992; 267:18210–18217. [ Links ]
68. Stadtman ER. Metal ion–catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic Biol Med 1990; 9:315–325. [ Links ]
69. Halliwell B, Gutteridge JMC. Lipid peroxidation, oxygen radicals, cell damage, and antioxidant therapy. Lancet 1984; 1:1396–1397. [ Links ]
70. Yatin SM, Aksenova M, Aksenov M, Butterfield DA. Effect of transglutaminase on Aβ (1–40) fibril formation and neurotoxicity. Alzheimer's Rep 1999; 2:165–170. [ Links ]
71. Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, et al. The Aβ peptide of Alzheimer's disease directly produces hydrogen peroxide thought metal ion reduction. Biochemistry 1999; 38:7609–7616. [ Links ]
72. Curtain CC, Ali F, Volitakis I, Cherny RA, Norton RS, Beyreuther K, et al. Alzheimer's disease amyloid–β binds copper and zinc to generate an allosterically ordered membrane–penetrating structure containing superoxide dismutasa–like subunits. J Biol Chem 2001; 276:20466–20473. [ Links ]
73. Mattson MP, Mattson EP. Amyloid peptide enhances nail rusting: novel insight into mechanisms of aging and Alzheimer's disease. Ageing Res Rev 2002; 1:327–330. [ Links ]
74. Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. Conservation of the sequence of the Alzheimer's disease amyloid peptide in dog, polar bear and five other mammals by cross–species polymerase chain reaction analysis. Brain Res 1991; 10:299–305. [ Links ]
75. Walter J, Grunberg J, Capell A, Pesold B, Schindzielorz A, Citron M, et al. Proteolytic processing of the Alzheimer disease–associated presenilin–1 generates an in vivo substrate for protein kinase C. Proc Natl Acad Sci USA 1997; 94:5349–5354. [ Links ]
76. Varadarajan S, Yatin SM, Aksenova M, Butterfield DA. Alzheimer's amyloid β–peptide–associated free radical oxidative stress and neurotoxicity. J Struct Biol 2000; 130:184–208. [ Links ]
77. Butterfield DA, Varadarajan S, Aksenova M, Link C, Yatin SM. On methionine and Alzheimer's amyloid β–peptide (1–42)–induced oxidative stress. Neurobiol Aging 1999; 20:339–342. [ Links ]
78. Vogt W. Oxidation of methionyl residues in proteins. Tools, targets, and reversal. Free Rad Biol Med 1995;18:93–105. [ Links ]
79. Naslund J, Schierhorn A, Hellman U, Lanfelt L, Roses AD, Tjernberg LO, et al. Relative abundance of Alzheimer Aβ amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci USA 1994; 91:8378–382. [ Links ]
80. Dado GP, Gellman SH. Redox control of secondary structure in a designed peptide. J Am Chem Soc 1994; 115:12609–12610. [ Links ]
81. Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine, 3th Edition. Oxford University Press, Oxford. 1999. [ Links ]
82. Zentella M, Saldaña Y. Papel fisiológico de los radicales libres. Boletín de Educación Bioquímica 1996; 15:1 52–161. [ Links ]
83. Halliwell B. Free radicals and antioxidants: a personal view. Nutr Rev 1994; 52:253–265. [ Links ]
84. Lledías F, Hansberg W. Catalase modification as a marker for singlet oxygen. Methods Enzymol 2000; 319:110–119. [ Links ]
85. Gutteridge JM. Hydroxyl radicals, iron, oxidative stress, and neurodegeneration. Ann NY Acad Sci 1994; 738:201–213. [ Links ]
86. Hansberg W. 2002. Biología de las especies de oxígeno reactivas. Mensaje bioquímico. XXVI. Facultad de Medicina, UNAM. México. [ Links ]
87. Halliwell B, Gutteridge JMC, Cross CE. Free radicals, antioxidants, and human disease: where are we now? J Lab Clin Med 1992; 119:598–620. [ Links ]
88. Butterfield DA. Amyloid β–peptide (1–42)–induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. Free Rad Res 2002; 36:1307–1313. [ Links ]
89. Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. β–amyloid–mediated vasoactivity and vascular endothelial damage. Nature 1996; 380:168–171. 119. [ Links ]
90. Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994; 77: 817–827. [ Links ]
91. Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature 1995; 378:776–779. [ Links ]
92. Smith MA, Harris PLR, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox–generated free radicals. Proc Natl Acad Sci USA 1997; 94:9866–9868. [ Links ]
93. Oteiza PI. A mechanism for the stimulatory effect of aluminum on iron– induced lipid peroxidation. Arch Biochem Biophys 1994; 308:374–379. [ Links ]
94. Cras P, Kawai M, Siedlak S, Mulvihill P, Grambetti P, Lowery D, et al. Neuronal and microglial involvement in beta–amyloid protein deposition in Alzheimer's disease. Am J Pathol 1990;137:241–246. [ Links ]
95. Colton CA, Gilbert DL. Production of superoxide anions by CNS macrophage, the microglia. FEBS Letters 1987; 223:284–288. [ Links ]
96. Smith MA, Harris PLR, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite–mediated damage in Alzheimer's disease. J Neurosci 1997; 17:2653–7. [ Links ]
97. Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, et al. Non–enzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta–peptide. Nat Med 1995; 1:693–699. [ Links ]
98. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A et al. RAGE and amyloid –β peptide neurotoxicity in Alzheimer's disease. Nature 1996;382:685–6891. [ Links ]
99. Butterfield DA, Stadtman ER. Protein oxidation processes in aging brain. Adv Cell Aging Gerontol 1997; 2:161–191. [ Links ]
100. Ledesma MD, Bonay P, Colaco C, Avila J. Analysis of microtubule–associated protein tau glycation in paired helical filaments. J Biol Chem 1994; 269:2161 4–21619. [ Links ]
101. Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, et al. Oxidative damage in Alzheimer's. Nature 1996;382:120–121. [ Links ]
102. Hensley K, Carney JM, Mattson MP, Aksenova M, Harrris M, Wu JF, et al. A model for beta–amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA 1994; 91:3270–3274. [ Links ]
103. Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology 1993; 43:1609–1611. [ Links ]
104. Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J. Nonsteroidal anti–inflammatory drugs in Alzheimer's disease. Neurology 1995; 45:51–55. [ Links ]
105. Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology 1997; 48:626–632. [ Links ]
106. Marzolo MP, von Bernhardi R, Inestrosa NC. Mannose receptor is present in a functional state in rat microglial cells. J Neurosci Res. 1999; 58:387– 395. [ Links ]
107. Giulian D. Ameboid microglia as effectors of inflammation in the central nervous system. J Neurosci Res 1987; 18:155–171. [ Links ]
108. Della Bianca V, Dusi S, Bianchini E, Dal Pr I, Rossi F. β–amyloid activates the O2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem 1999; 274:15493–15499. [ Links ]
109. Klegeris A, Walkner DG, McGeer PL. Activation of macrophages by Alzheimer β–amyloid peptide. Biochem Biophys Res Commun 1994; 199:984–991. [ Links ]
110. Hurst JK, Barrette WC. Leukocytic oxygen activation and microbicidal oxidative toxins. Crit Rev Biochem Mol Bull 1989; 24: 271–328. [ Links ]
111. Meda L, Cassatella MA, Szendrei G, Otvos L, Baron P, Villalba M, et al. Activation of microglial cells by Aβ protein and interferon–gamma. Nature 1995; 374:647–650. [ Links ]
112. Rogers J, Lue LF. Microglial chemotaxis, activation, and phagocytosis of amyloid beta–peptide as linked phenomena in Alzheimer's disease. Neurochem Int 2001; 39:333–340. [ Links ]
113. Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 1995; 38:357–366. [ Links ]
114. Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid β–peptide–role of the lipid peroxidation product 4–hydroxynonenal. J Neurochem 1997; 69:273–284. [ Links ]
115. Blum–Degen D, Frolich L, Hoyer S, Riederer P. Altered regulation of brain glucose metabolism as a cause of neurodegenerative disorders. J Neural Transm 1995; S139–S147. [ Links ]
116. Schubert D, Behl C, Lesley R, Brack A, Dargusch R, Sagara Y, Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci USA 1995; 92:1989–1993. [ Links ]
117. Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, et al., Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol. 2001; 171:29–45. [ Links ]
118. Hadding A, Kaltschmidt B, Kaltschmidt C. Overexpression of receptor of advanced glycation end products hypersensitizes cells for amyloid beta peptide–induced cell death. Biochim Biophys Acta 2004;1691:67–72. [ Links ]
119. Liu Y, Dargusch R, Schubert D. β–amyloid toxicity does not require RAGE protein. Biochem Biophys Res Commun. 1997; 128:238–246. [ Links ]
120. Hussain MM, Maxfield FR, Mas–Oliva J, Tabas I, Ji ZS, Innerarity TL, Mahley RW. Clearance of chylomicron remnants by the low density lipoprotein receptor–related protein/ β2–macroglobulin receptor. J Biol Chem 1991; 266:1 3936–1 3940. [ Links ]
121. Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RC, et al. β2–macroglobulin is a genetically associated with Alzheimer's disease. Nat Genet 1998; 19:357–360. [ Links ]
122. Knauer MF, Orlando RA, Glabe CG. Cell surface APP751 forms complexes with protease nexin 2 ligands and is internalized via the low density lipoprotein receptor–related protein (LRP). Brain Res 1996;740:6–14. [ Links ]
123. Marzolo MP, von Bernhardi R, Bu G, Inestrosa NC. Expression of β2–macroglubulin receptor/LRP in rat microglial cells. J Neurosci Res 2000; 60:401–411. [ Links ]
124. Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, et al. Glycoprotein 330/megalin: probable role in receptor–mediated transport of apolipoprotein J alone and in a complex with Alzheimer's disease amyloid–β at the blood–brain and blood–cerebrospinal fluid barriers. Proc Natl Acad Sci USA 1996; 93:4229–434. [ Links ]
125. Paresce DM, Ghosh RN, Maxfield FR. Microglial cell internalize aggregates of Alzheimer's disease amyloid β–protein via a scavenger receptor. Neuron 1996;17:553–565. [ Links ]
126. El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor–mediated adhesion of microglia to β–amyloid fibrils. Nature 1996; 382:716–719. [ Links ]
127. Christie RH, Freeman M, Hyman BT. Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer's disease. Am J Pathol 1996; 148:399–403. [ Links ]
128. Krieger M. The other side of scavenger receptors: pattern recognition for host defense. Curr Opin Lipid 1997; 8:275–280. [ Links ]
129. Rohrer L, Freeman M, Kodama T, Penman M, Krieger M. Coiled–coil fibrous domains mediate ligand binding by macrophage scavenger receptor type II. Nature 1990; 343:570–572. [ Links ]
130. Guaderrama–Díaz M, Solis CF, Velasco–Loyden G, Laclette JP, Mas–Oliva J. Control of scavenger receptor–mediated endocitosis by novel ligand of different lenght. Mol Cell Biochem 2005; 271:123–132. [ Links ]
131. Mas–Oliva J, Velasco–Loyden G, Haines TH. Receptor pattern formation as a signal for the capture of lipoproteins. Biochem Biophys Res Commun 1996; 224:212–218. [ Links ]
132. Hampton RY, Golenbock DT, Penman M, Krieger M, Raetz CR. Recognition and plasma clearance of endotoxin by scavenger receptors. Nature 1991; 352:342–344. [ Links ]
133. Honda M, Akiyama H, Yamada Y, Kondo H, Kawabe Y, Takeya M, et al. Immunohistochemical evidence for a macrophage scavenger receptor in Mato cells and reactive microglia of ischemia and Alzheimer's disease. Biochem Biophys Res Commun 1998; 245:734–740. [ Links ]
134. Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid beta–protein by microglial cells. J Biol Chem 1997; 272:29390–29397. [ Links ]
135. Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta–peptide by microglial cells. J Biol Chem 1999; 274: 32301–32308. [ Links ]
136. Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, et al. Aβ–induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol 2001; 158:63–73. [ Links ]
137. Husemann J, Loike JD, Kodama T, Silverstein SC. Scavenger receptor class B type I (SR–BI) mediates adhesion of neonatal murine microglia to fibrillar β–amyloid. J Neuroimmunol 2001; 114:142–150. [ Links ]
138. Husemann J, Silverstein SC. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer's disease brain. Am J Pathol 2001; 158:825–832. [ Links ]
139. Bamberger MA, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β–amyloid mediates microglial activation. J Neurosci 2003; 23:2665–2674. [ Links ]
140. Wyss–Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid–beta in vitro and in situ Nat Med 2003; 9:453–457. [ Links ]
141. El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. Microglia, scavenger receptors, and the pathogenesis of Alzheimer's disease. Neurobiol Aging 1998;19:S81–S84. [ Links ]
142. Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, et al. Specific domains of beta–amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci 1996; 16:6021 –6037. [ Links ]
143. Aguilar–Gaytán R, Mas–Oliva J. Oxidative stress impairs endocytosis of the scavenger receptor class A. Biochem Biophys Res Commun 2003; 305:510–517. [ Links ]
144. Manzano–León N, Guaderrama–Díaz M, Mas–Oliva J. Efecto del estrés oxidativo sobre la función del receptor scavenger. Memorias del XIV Congreso de Bioenergética y Biomembranas. Sociedad Mexicana de Bioquímica. 2005.p. 56–63. [ Links ]
Avenida Cuauhtémoc 330, 1er. piso,
México, D.F., 06725, México

gacetamx@starnet.net.mx
postado por Blog do Paim @ 14:09
0 Comentários
![]()



0 Comentários:
Postar um comentário
Assinar Postar comentários [Atom]
<< Página inicial